
grammar2vec: Molecular property prediction using SMILES grammar
Published:
Ever wondered how to inject chemistry knowledge into machine learning models to accurately predict properties of molecules? How would a molecule be represented before it is input to an ML model? How could the representation be made more chemistry-aware and ensure it is complete and accurate? Do natural language processing methods have anything to do with such representations?
We developed a framework called grammar2vec (shown in Figure 1), similar to word2vec for words -- that I'm sure most of us are familiar with thanks to the famous King - Man + Woman = Queen example. Word2vec is an unsupervised representation learning approach that learns dense vectors for representing words that also capture their underlying semantics and meaning to a large extent. Grammar2vec is the molecular equivalent of word2vec and Grammar2vec generates a high dimensional numeric vector representation for molecules. These numeric molecular representations are similarly learnt in an unsupervised manner, while capturing their semantics to a large extent. A natural question that arises then is -- how to represent the molecule textually as the basis for generating representation using grammar2vec, a framework similar to word2vec for natural language text?
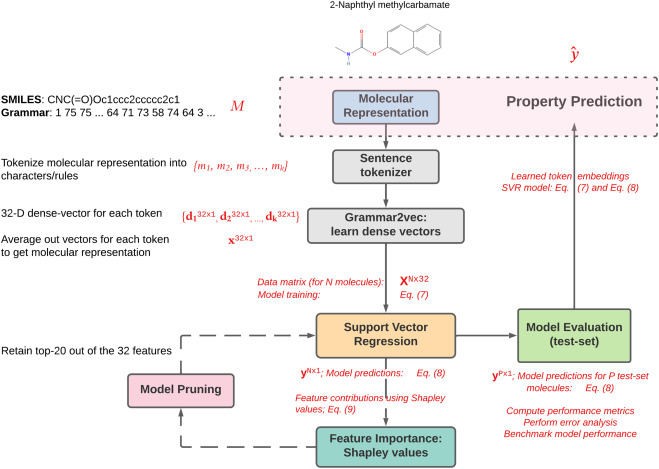
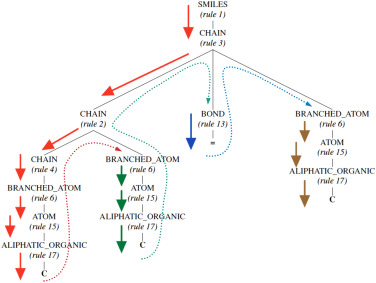
To answer this question, let's look at a common text-based representation of molecules called the SMILES representation. For instance, for say propene, the SMILES representation would be 'CC=C' where each 'C' indicates the presence of an aliphatic carbon atom and '=' indicates the presence of a double-bond between two C atoms. While these representations are great in representing underlying chemistry succinctly, they miss out on explicit structural information such as the nature of chains, type of bonds and atoms, and so on. To explicitly include such structural information in a molecule's text-based representation, we combine the SMILES representation with the underliyng SMILES grammar. We parse the SMILES string using its underlying grammar to assign a hierarchical structure to the SMILES representation, called a grammar parse-tree. In this grammar parse-tree, the intermediate nodes provide useful chemistry-based structural meta information. For instance, the same propene molecule would be represented as shown in the hierarchical tree in Figure 2. And when this molecular grammar tree is traversed in a depth-first manner, we get a sequential representation that is an inline text-based representation just like SMILES string but also contains the intermediate structural information embedded in the parse tree. This sequential grammar-based representation for propene would be "1, 3, 2, 4, 6, 15, 17, 6, 15, 17, 13, 6, 15, 17", comprising a sequence of production rules at each step/node in the SMILES grammar parse-tree. Therefore, using the sequential grammar representation, we generate dense vector embeddings using a word2vec-like framework that are used to represent a given molecule, irrespective of the property that needs to be predicted. Details on the implementation could be found in our published article.
The dense molecular representations are used as features for a given ML model with the property of interest to be predicted as the target variable. This allows for any suitable model depending on the dataset size, problem formulation, and target property of interest to be used with grammar2vec-based molecular descriptors. Moreover, we demonstrated that the models built using these features are chemistry-aware in the sense that correlated properties have correlated feature importances. The model performance was benchmarked on boiling point and critical temperature datasets, and we observed that the test-set R-squared values (a measure of goodness of fit for regression models) were 0.91 and 0.90, respectiely. Detailed results are shown below in Figure 3.
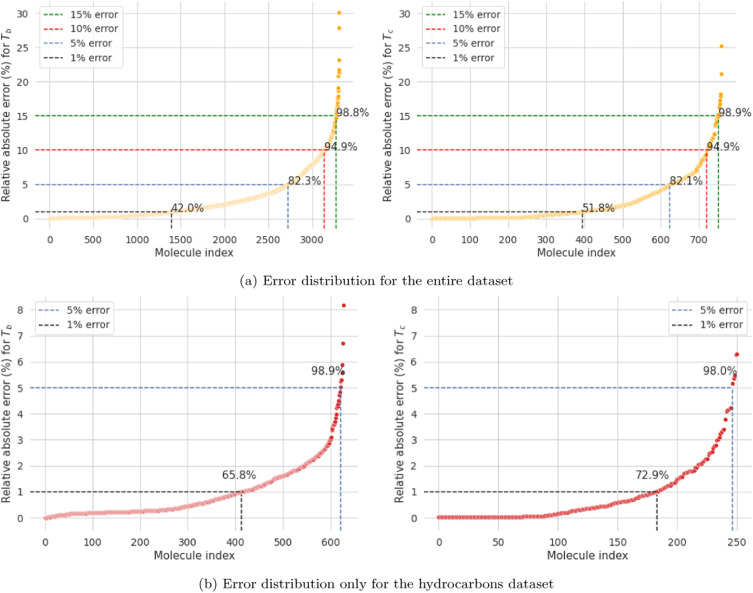
We observe that ~99% of all the predictions fall within a 15% error threshold and the maximum prediction errors are capped at 30% and 25% for the boiling point and critical temperature predictions. Moroever, the performance on the hydrocarbons dataset is significantly better with nearly 98-99% of all the molecules within an error-threshold of less than 1%. The gradually increasing trend in the relative errors (as opposed to a steep increase beyond a certain number of molecules), highlights a smooth functional mapping from the features to the property values learned by the ML-based regression model. As a consequence of this, the model has better generalizability. Therefore, we infer that our model is robust towards encountering new molecules or the presence of outliers in the dataset. The latter is a major issue because new molecules are being synthesized at a much faster pace and the experimentally measured properties dataset often have errors and outliers.
In summary, the Grammar2vec framework could be used for estimating several other molecular properties using relatively simple and interpretable machine learning models owing to the rich molecular descriptors learnt using grammar2vec. We envision that such representations that are rooted in chemistry would be of significant value not only for the thermodynamic property prediction task but also for other chemistry problems requiring data-driven modeling. Further details could be found in our published article.
